Our recent newsletter Feature Article highlighted ISO 13485 audits applicable to the medical device industry. A common question we receive in relation to regulatory requirements in the U.S. is the relationship to FDA 21 CFR Part 820.
To ensure medical devices are safe and effective, the U.S. Food and Drug Administration regulates current good manufacturing processes (CGMP’s) also known as FDA 21 CFR Part 820. FDA’s Quality System Regulation Part 820 is harmonized with ISO 13485:1996, which is based on ISO 9001:1994.
According to the FDA,
“The QS regulation applies to finished device manufacturers who intend to commercially distribute medical devices. A finished device is defined in 21 CFR 820.3(l) as any device or accessory to any device that is suitable for use or capable of functioning, whether or not it is packaged, labeled, or sterilized. Because the regulation must apply to so many different types of devices, the regulation does not prescribe in detail how a manufacturer must produce a specific device. Rather, the regulation provides the framework that all manufacturers must follow by requiring that manufacturers develop and follow procedures and fill in the details that are appropriate to a given device according to the current state-of-the-art manufacturing for that specific device.” CFR – Code of Federal Regulations Title 21In Section 820.22 of the Code, a quality audit is prescribed.
“Each manufacturer shall establish procedures for quality audits and conduct such audits to assure that the quality system is in compliance with the established quality system requirements and to determine the effectiveness of the quality system. Quality audits shall be conducted by individuals who do not have direct responsibility for the matters being audited. Corrective action(s), including a reaudit of deficient matters, shall be taken when necessary. A report of the results of each quality audit, and reaudit(s) where taken, shall be made and such reports shall be reviewed by management having responsibility for the matters audited. The dates and results of quality audits and reaudits shall be documented.”Pro QC performs FDA 21 CFR Part 820 audits that incorporate an evaluation of each of the following regulation components:
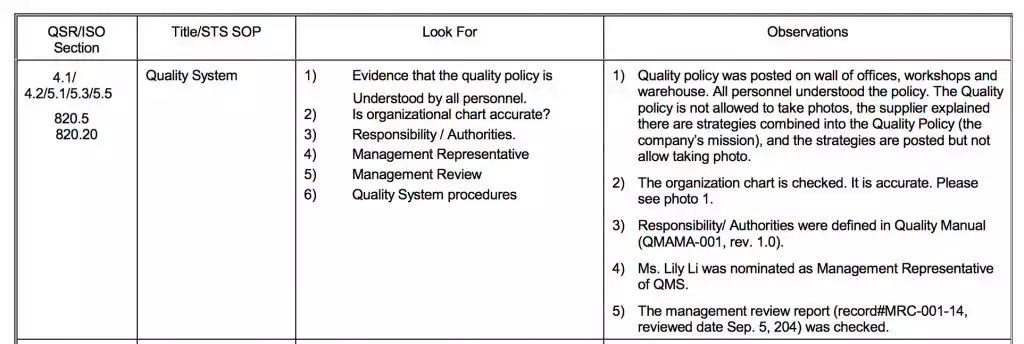
- Quality System
- Personnel
- Document Control
- Records Control
- Device Master Record (DMR)
- Device History Record (DHR)
- Contract Review
- Design Controls
- Purchasing Controls
- Control of Customer Supplied Product
- Identification Traceability
- Production & Process Controls
- Installation
- Process Validation
- Receiving, In-Process and Finished Device Acceptance
- Inspection, Measuring & Test Equipment
- Device Labeling
- Non-Conforming Product (NCP)
- Corrective & Preventative Action
- Device Packaging, Handling & Storage Distribution
- Statistical Techniques
Contact us to review an example report, or further discuss how we meet the growing needs of the medical device industry.


